




















引言
膠質瘤是成人及兒童腦部原發腫瘤中最常見的瘤種。對該組腫瘤的分子研究,已大大提高了我們的診斷準確性以及預后判斷能力。近期我們將根據《Surg Pathol Clin》雜志綜述文章,為大家編譯介紹膠質瘤分子病理相關知識點。
關鍵詞
IDH1/2 突變 ATRX 突變 組蛋白 H3 K27M 突變 1p19q 丟失 BRAF 融合 BRAF V600E 突變
提要
異檸檬酸脫氫酶 1/2 (IDH1/2) 突變狀態和 1p19q 染色體臂狀態對于準確分類成人彌漫性膠質瘤至關重要。
IDH1/2 突變的腫瘤比野生型腫瘤具有顯著更好的結果。
在缺乏診斷性組織學特征的情況下,膠質母細胞瘤的診斷可以僅基于特征性分子特征。
高級別小兒神經膠質瘤的特征是具有 K27M 或 G34 突變的組蛋白 H3 突變。
兒科低級別神經膠質瘤最常通過 BRAF 基因融合或點突變顯示 MAPK 通路改變。

概述
70歲以下人群中,中樞神經系統原發腫瘤僅占原發癌的2%,但在因癌導致的死亡中所占比例卻為7%。0-14歲兒童中,腦腫瘤是第二常見的癌癥類型,且兒童癌相關死亡中的首要原因。腦原發腫瘤中膠質瘤約占所有中樞神經系統原發腫瘤的26%。生物學行為方面,膠質瘤的差異較大,自相對良性、緩慢生長、境界清楚至快速侵襲性生長、彌漫浸潤性表現均可。
膠質瘤,即起源于中樞神經系統膠質細胞的腫瘤。目前來說,其分類還主要是根據組織病理學表現,即瘤細胞像哪種對應的正常成分,具體分為星形細胞瘤、少突膠質細胞瘤。不過,分子病理的介入已顯著影響了病理層面對該組瘤種的認識。具體如:IDH1/2突變、且有ATRX及TP53突變,是彌漫性星形細胞瘤的特征;IDH1/2突變、且有1p19q缺失,是少突膠質細胞瘤的特征;受體酪氨酸激酶基因局灶擴增、TERT啟動子突變、10號及13號染色體缺失且7號染色體三體是膠質母細胞瘤的特征,可用于診斷;低級別膠質瘤中有BRAF基因的融合及突變,高級別膠質瘤中有組蛋白H3的突變,也可用于診斷。
進行分子檢測鑒別膠質瘤之前,一般要結合臨床及影像學特征。因為相比其他癌種來說,腦腫瘤的年齡、部位、分子遺傳學特征方面有顯著相關性,膠質瘤也不例外。小兒的彌漫性膠質瘤多位于中線部位,且有組蛋白H3的K27M突變;相反,較為良性的兒童膠質瘤是由MAPK通路改變所致、最常見為BRAF基因融合或點突變,且位于小腦或視路上。年齡較大兒童發生的惡性半球膠質瘤具有組蛋白H3 G34突變,而生長緩慢的皮質腫瘤多為BRAF突變所致、且為境界清楚的生長方式。年輕成人發生的彌漫性星形細胞瘤分子特征表現為三打擊,即IDH1/2、TP53、ATRX突變,且以精準的順序出現。相反,IDH1/2突變、并有染色體1p19q缺失,則是少突膠質細胞瘤的特征。少突膠質細胞瘤最常見于額葉,生長緩慢并有鈣化。60歲以上的年齡較大成人中,腫瘤為IDH野生型彌漫性膠質瘤,主要是由拷貝數改變、受體酪氨酸激酶及PI3K/PTEN/AKT和Rb信號通路改變所致。IDH突變型及IDH野生型腫瘤在臨床表現方面也存在顯著差異:IDH突變型腫瘤更常見癲癇,出現異常出血的幾率低;而IDH野生型彌漫性膠質瘤發生血栓栓塞并發癥的幾率較高。
分子病理特征
IDH突變的彌漫型膠質瘤
IDH1/2突變的發現,帶來了對膠質瘤生物學行為理解的進步,也改變了彌漫型膠質瘤的傳統分級方案。IDH1/2突變的星形細胞瘤更多見于較年輕的成人,相同分級情況下要比IDH野生型患者的預后更好。
研究表明,IDH1/2突變膠質瘤中,ATRX和TP53突變之間有顯著相關性。此外,研究表明MGMT啟動子甲基化與IDH1和TP53突變有關。
IDH突變型和IDH野生型腫瘤屬于不同的病種,需不同于以往組織學分級方案的其他分級。為中樞神經系統腫瘤分類提供分子信息及實踐方法委員會(Consortium to Inform Molecular and Practical Approaches to CNS Tumor Taxomomy)(非官方的世界衛生組織)工作中整合相關分子特征,提出了一個整合分類方案,將IDH突變型星形細胞瘤分為:
- 星形細胞瘤,IDH突變型,WHO分級2級:無間變性表現,核分裂活性低,無微血管增生或壞死;
- 星形細胞瘤,IDH突變型,WHO分級3級:有間變性表現,核分裂顯著,無微血管增生或壞死;
- 星形細胞瘤,IDH突變型,WHO分級4級:有微血管增生或壞死,或有CDKN2A/B的純合型缺失。
CDKN2A/B純合型缺失整合入4級的分類,是由于這一特征與IDH突變型星形細胞瘤患者生存期縮短有顯著相關性。該建議中對IDH突變型IV級腫瘤并未保留膠質母細胞瘤的名稱。
少突膠質細胞瘤是彌漫型膠質瘤的一部分,主要見于50-60歲患者的大腦兩半球。世界衛生組織將其分為2級或3級腫瘤。研究表明,少突膠質細胞瘤發生過程中的初始事件之一就是IDH1/2突變,甚至在1p/19q的缺失之前。確定IDH1或IDH2突變后,同時有1p/19q的缺失,是2016年版世界衛生組織分類中診斷少突膠質細胞瘤的要求。1p/19q的共缺失可能與FUBP1及CIC同時存在失活突變有關,二者是分別位于1p和19q的腫瘤抑制基因。FUBP1和CIC同時存在突變,是少突膠質細胞瘤所特有,在其他中樞神經系統腫瘤中罕見。少突膠質細胞瘤中的其他突變還有:約80%的腫瘤存在TERT啟動子突變,且與ATRX突變互斥存在。
1p19q的缺失是少突膠質細胞瘤的定義性特征。大部分腫瘤缺失的是1q的兩條染色體臂、1p的1條染色體臂、19p的兩條染色體臂、19q的一條染色體臂,部分腫瘤會同時有伴多體性的缺失,且1p:1q和19p:19q的比例不一(原文如此,譯者注)。這種相對的缺失或超級缺失(supeloss)與腫瘤的快速進展有關。1p/19q的缺失同時伴多倍體與不良預后、病變發生時候年齡較輕、組織學分級較高、無進展生存和總生存較差有關。最近有納入333例的大規模多機構研究證實多倍體對預后有不利影響。少突膠質細胞瘤中CDKN2A/B缺失及PIK3CA改變也偶見于高級別腫瘤。
IDH1/2與星形細胞瘤中的ATRX/TP53突變、或與少突膠質細胞瘤中1p19q缺失有顯著相關,導致無法分類的彌漫性膠質瘤數量顯著減少。不過,少突膠質細胞瘤一般很少需分子檢測支持,一般不建議進行分子檢測。
免疫組化可用于檢測IDH1 R132H突變,通過突變特異性抗體,IDH突變型膠質瘤中陽性率為80-90%。其他IDH1/2突變較為少見,但具有同等重要的預后意義,需通過基因測序檢出。免疫組化還可用于檢出ATRX表達缺失,且p53強陽性著色可提示TP53突變;不過,常有的抗體并非突變特異性,可能還需分子檢測證實。
1p和19q的狀態評估是各實驗室公認的檢測,FISH、PCR檢測雜合性缺失、二代測序、基因分型、甲基化分析等,都可以提供1p19q的狀態。需要注意的是,分析1p9q的染色體臂時,評估的是染色體1p和19q的共同缺失,因為單一的19q缺失常見于星形細胞瘤,1p的局灶缺失常見于IDH野生型膠質母細胞瘤。
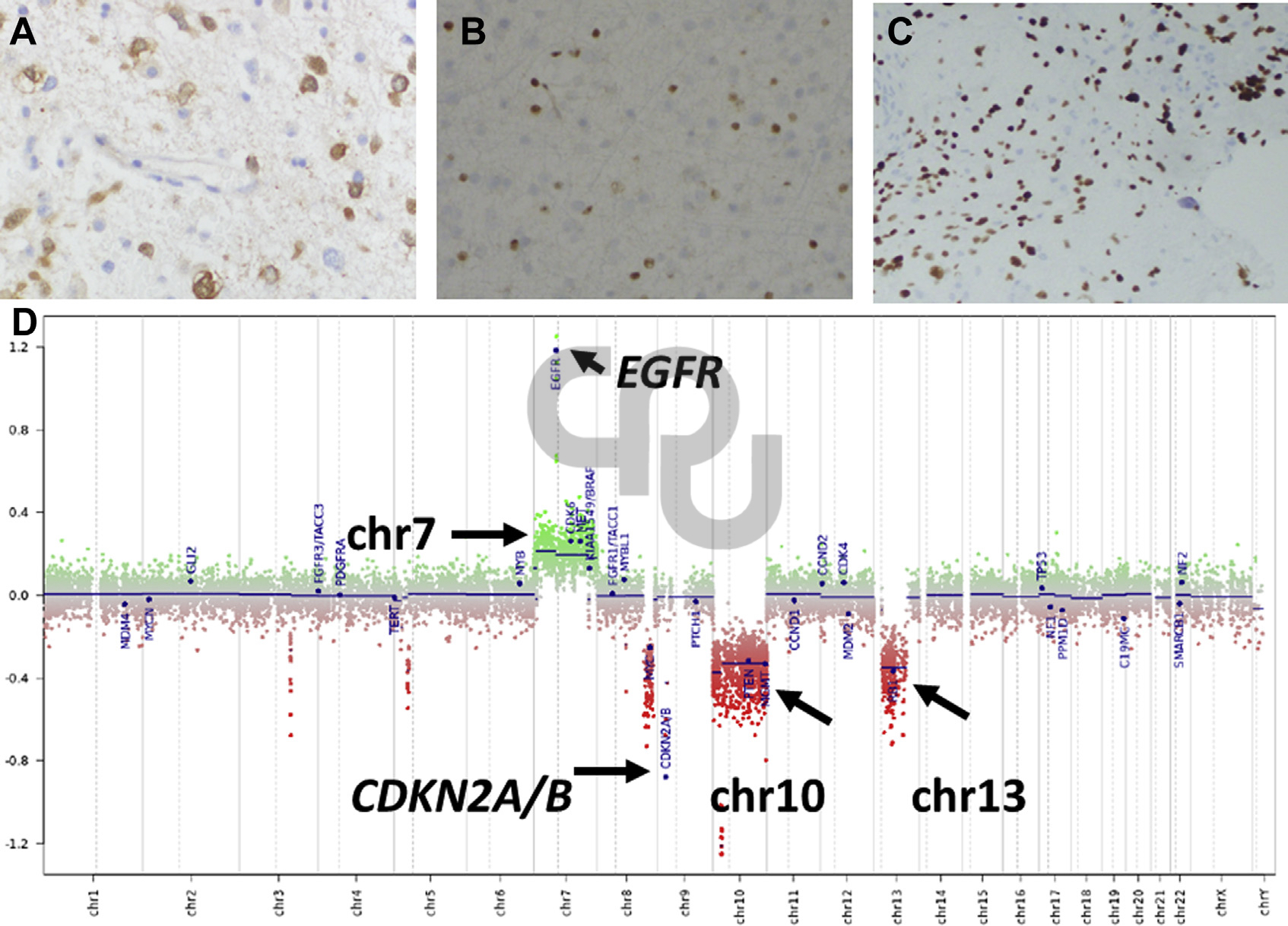
圖1. 低級別和高級別星形細胞瘤的分子特征:(A)IDH1 R132H突變特異性抗體免疫組化,瘤細胞呈強陽性表達;(B)ATRX免疫組化,浸潤性膠質瘤細胞中著色缺失,正常膠質細胞和神經元中仍有著色;(C)p53免疫組化,細胞核強陽性著色,提示有突變。這一三聯表現是IDH突變型膠質瘤的特征。(D)IDH野生型星形細胞瘤、膠質母細胞瘤的分子特征,包括7號染色體三體并有EGFR的局灶擴增、CDKN2A/B的純合型缺失、10號及13號染色體單體。存在這些分子特征的勤快下,即使無高級別組織學特征,也應診斷膠質母細胞瘤。

圖2. 少突膠質細胞瘤的分子特征。(A)少突膠質細胞瘤的特征是IDH1/2突變,可通過突變特異性抗體做免疫組化來診斷;此外,所有的少突膠質細胞瘤必須同時存在1p(B)及19q(C)的染色體缺失,可通過FISH或其他方法檢測出來,如全基因組測序(D)。
未完待續
參考文獻
Galbraith K, Snuderl M. Molecular Pathology of Gliomas. Surg Pathol Clin. 2021;14(3):379-386.
doi:10.1016/j.path.2021.05.003